手性非天然氨基酸是许多药物分子和天然产物的合成砌块,将非天然氨基酸结构片段引入医药分子和天然产物中,能够改善药物的理化性质以及药代动力学特性,提高其成药性。但是非天然氨基酸结构多样,且具有多个手性中心,其传统合成工艺十分复杂、反应步骤多、收率低、立体选择性差。镍螯合物诱导不对称合成方法已成为该类化合物高效高立体选择性合成的重要策略之一。
中国科学院上海药物研究所科研人员一直致力于镍螯合物诱导手性非天然α-和β-氨基酸的研究工作。运用“镍螯合物诱导不对称合成非天然氨基酸”的方法学研究策略,成功构建并合成了一系列结构多样、种类丰富的非天然手性氨基酸化合物库,为药物活性化合物的快速制备打下了坚实的化学基础。
在成功制备α,β-二氨基氨基酸、3-氨基天冬氨酸、4-取代谷氨酸、含三氟甲基链状氨基酸、β-取代-α,γ-二氨基氨基酸以及β-取代色氨酸的基础上,引入C-H活化反应研究策略,成功利用杂环偶联反应制备3-吲哚甘氨酸及其衍生物(Chem. Commun. 2013, 49, 2575-2577),运用交叉脱氢偶联(CDC)反应构建四氢异喹啉-1-甘氨酸及其衍生物(J. Org. Chem. 2013, 78, 11204−11212)。该系列研究成果的发表,引起了国内外同行的广泛关注和好评,分别应Current Pharmaceutical Design 和 Chimia 杂志主编的邀请,先后为其撰写镍螯合物诱导不对称合成氨基酸的研究综述(Curr. Pharma. Design 2010, 16, 1252-1259.和Chimia 2011, 65, 919-924.),其中第二篇被选为当期杂志(Chemistry in China)的封面文章。
传统的β-氨基酸合成方法工艺复杂、合成的β-氨基酸结构单一,不能满足生物医药领域对β-氨基酸结构多样性的需求。科研人员在前期研究工作的基础上,首次开发了“一锅两步法”制备β-氨基酸等当体镍螯合物(J. Org. Chem. 2007, 72, 8932-8934.),并通过将该螯合物与卤代试剂进行烷基化反应,成功构建了α,α-双取代-β-氨基酸类衍生物(J. Org. Chem. 2010, 75, 1717-1722.)。另外,通过Suzuki偶联反应和Diels-Alder反应,成功构建了α-取代β2-氨基酸(J. Org. Chem. 2011, 76, 6649-6656.)以及含有多个手性中心的环状β-氨基酸类衍生物(Amino Acids 2013, 44, 791-796.)。
为了大规模制备手性非天然β-取代β-氨基酸,该课题组近期首次实现N,C-无保护的β-氨基酸化学拆分方法。此方法突破以往利用镍螯合物氨基酸等当体不对称合成非天然氨基酸的研究思路,设计了结构新颖、通用易得的手性配体,在金属镍的介导下与消旋的β-氨基酸中构型匹配的氨基酸发生高选择性的螯合反应,再通过简单水解释放对映纯的β-氨基酸,从而实现了对消旋β-氨基酸的手性拆分。该方法填补了化学拆分N,C-无保护的β-氨基酸领域的空白,为工业上大规模制备手性非天然β-取代β-氨基酸提供了新方法。除此之外,此方法具有操作简单、立体化学选择性强 (dr > 99:1)、底物使用范围广、手性配体可回收、易于工业生产等优点。同时,研究人员还将此方法应用于抗糖尿病药物西他列汀的不对称合成。相关研究成果发表在Angewandte Chmie 杂志上(Angew. Chem. Int. Ed. 2014, 53, 7883 –7886)。
该系列研究工作得到了国家自然科学基金委员会的大力支持。
文章链接:1 2 3 4 5 6 7 8
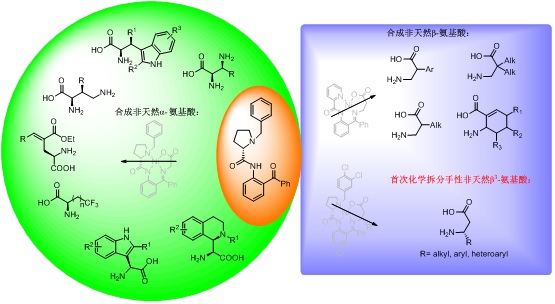
通过不同配体的镍螯合物化学合成、拆分手性非天然α-和β-氨基酸

 联系我们
联系我们